High-quality wallflower genome reveals its chromosome evolution and flower color variation
Recently we published a near telomere-to-telomere reference genome of Erysimum cheiri in Plant Physiology (Chen et al., 2026). We used the genome to investigate the origin of Erysimum karyotypes and to determine how its flower color variation is determined. We mapped the metabolic pathways for carotenoids and flavonoids, identifying the hub genes regulating their biosynthesis. This is the first chromosome-level reference genome for the important ornamental species, providing molecular insights into flower color variation.
Wallflower was chosen for the cover!
The paper was featured by News and Views.
The whole lab went out to celebrate the St. Martin's Day (November 11) and the very first wines of the new vintage!

BSc/MSc thesis topic - sequence and build genome, solve mysteries. Looking for a motivated student(s) to sequence, assemble, and analyze plant genomes!

Dr. Yile Huang successfully defended her PhD thesis!
On October 16, Yile successfully defended her PhD thesis entitled Genomics of chromosomal dysploidy in plants. Her primary focus was on post-polyploid genome diploidization in mesopolyploid crucifer genomes, which is often associated with descending dysploidy (the reduction of chromosome number). Congratulations, Dr. Huang :-)!
Terezie Malík Mandáková is among this year’s Top Czech Women Scientists selected by Forbes
Congratulations, Teri! More info (in Czech) can be found here: https://forbes.cz/top-vedkyne-ceska-2025/
The largest genome to date in the Brassicaceae decoded!
Martin Lysak, together with the team from Huazhong Agricultural University in China (led by Xianhong Ge), coordinated the sequencing of the largest ever reported genome in the Brassicaceae family. The genome belongs to Matthiola incana, commonly known as stock, a fragrant ornamental species that is grown both as a cut flower and as a bedding plant. The genome of Matthiola (2 G) is twelve times larger than that of Arabidopsis, even though both species have a similar number of chromosomes. We have
succeeded in obtaining a high-quality chromosome-scale genome assembly of M. incana by integrating PacBio HiFi reads, Illumina short reads and Hi-C data. Our results suggest that the primary reason for genome obesity in M. incana is the massive expansion of long terminal repeat retrotransposons (LTR-RTs), particularly from the Angela, Athila and Retand families. The work was published in Plant Biotechnology Journal.
Chromosome fusion breakpoints elucidated phylogenomic relationships within the model family
Led by researchers from Sichuan University and Martin Lysak from CEITEC MUNI, the study published in Nature Communications examined chromosome fusion breakpoints as markers of phylogeny to determine evolutionary relationships within the mustard family (Brassicaceae). By comparing shared fusion breakpoints and their sequential order across the phylogenetic tree, the team resolved some long-standing controversies regarding the relationships within the two major lineages harbouring the model species Arabidopsis thaliana and Brassica crop species (e.g. canola, cabbage, broccoli).
Successful symposium at the International Botanical Congress in Madrid (July 21 to 27, 2024)
A two-session symposium on Advances in phylogenomics and systematics of the Brassicaceae was organized by Martin Lysak and Marcus Koch and sponsored by Annals of Botany. A total of 12 speakers discussed unprecedented advances in systematics, phylogenomics and evolutionary research in the Brassicaceae and shared their most interesting observations and findings with the audience. The opening lecture of the symposium was given by Dr. Ihsan Al-Shehbaz (Missouri Botanic Garden), the world’s leading authority on the taxonomy of the Brassicaceae family. Ihsan gave an amazing talk on the astonishing advances in the systematics and phylogenetics of the Brassicaceae over the last quarter century. Some of the papers presented will appear in the special issue of Annals of Botany, Advances in Crucifer Research in the -Omics Era.
MUNI SCIENTIST 2023 Award for the members of the Lysak Lab

The Grant Agency of Masaryk University presented the MUNI Scientist 2023 Awards. Martin Lysák, Terezie Mandáková, Xinyi Guo and Yile Huang were awarded for articles published in Plant Journal, Plant Journal, Frontiers in Plant Science and Nature Communication.
New global Brassicaceae phylogeny published!

An internationl team of researchers published an unprecedented phylogeny of the mustard family (Brassicaceae) with representatives from nearly all 349 genera. It is expected that these results will boost fundamental and applied plant biological research. This work was now published in Current Biology!
The rare Australian endemic species Carinavalva schizopetala grown in our laboratory made it to the cover! Seeds provided by Daniel Duval, image by Terezie Mandáková.
The horseradish genome decoded
Czech and Chinese researchers have obtained the first gap-free sequence of the horseradish genome. Combining data from Illumina, Oxford Nanopore Technology, PacBio HiFi, and Chromatin Conformation Capture (Hi-C) sequencing, they generated the first telomere-to-telomere (T2T) genome assembly of the allotetraploid horseradish genome (Armoracia rusticana). The genome sequence allowed the researchers to investigate the genetic basis of the biosynthesis of glucosinolates (GSLs) and horseradish peroxidases. While GSLs are precursors of isothiocyanates responsible for the characteristic pungent and spicy taste of horseradish, horseradish peroxidases are frequently used as a reporter enzyme in diagnostics and histochemistry. The first T2T assembly of the horseradish genome expands our understanding of polyploid genome evolution and provides a fundamental genetic resource for breeding and genetic improvement of horseradish. The study, led by Fei Shan (Beijing Academy of Agriculture and Forestry Sciences) and Martin Lysak (CEITEC Masaryk University), has now been published in Nature Communications.
Horseradish (Armoracia rusticana), which like cabbage or wasabi belongs to the mustard family, contains glucosinolates (GLSs) that break down into isothiocyanates with characteristic pungency. Horseradish is grown for its root, which, when grated, becomes pungent and is processed into spicy condiments. GLSs or their breakdown products provide numerous benefits to the human body, including anti-cancer, anti-inflammatory, and anti-microbial properties. Despite its increasing importance as a “super health food” limited genomic resources have prevented the application of advanced breeding techniques to horseradish and the elucidation of the genetic basis of its important traits.
The research team, led by Fei Shen from the Institute of Biotechnology Research, Beijing Academy of Agriculture and Forestry Sciences, and Martin Lysak from the Central European Institute of Technology (CEITEC), Masaryk University, reported the telomere-to-telomere (T2T) gap-free reference genome of allotetraploid horseradish sequenced using a comprehensive strategy. Analysis of the first T2T assembly of an allotetraploid genome (i.e., genome consisting of two parental subgenomes) in the mustard family demonstrated the role of transposable DNA elements (long terminal repeat retrotransposon) and DNA methylation in subgenome differentiation. Using advanced genomic techniques, scientists have fully characterized the epigenomic architecture and 3D chromatin structure of two horseradish subgenomes and deciphered the dramatic contribution of divergence and bursts of DNA repeats to subgenomic divergence. The in-depth analysis of the horseradish peroxidase family provides important resources for studying the full spectrum of horseradish isoenzymes. In addition, researchers have identified the genetic basis underlying the wide diversity of GSLs and their degradation products in horseradish. Allopolyploidization and continuous tandem duplications have allowed preferential retention of GSL genes, contributing to the wide diversity of GSLs and their degradation products in horseradish.
Overall, the first T2T assembly of the allotetraploid horseradish genome, together with its epigenomic architecture and 3D chromatin structure, expands our understanding of polyploid genome evolution and provides a fundamental genetic resource for breeding and genetic improvement of horseradish. The high-quality horseradish genome will be a valuable genomic resource for biologists, biochemists, and agricultural scientists.
The publication in Nature Communications is available also here.
Step into Africa! CEITEC scientists elucidate the role of ancient whole-genome duplications in the origin of South Africa's unique flora
Year after year, the brown desert landscape of Namaqualand in South Africa is transformed into one of the most beautiful floral displays in the world. From the end of the South African winter season into spring, thousands of daisies and flowers of all colors transform the arid region into a blooming carpet. Among other plants, species of the genus Heliophila (>100 species) from the mustard family contribute to the annual flowering miracle.
CEITEC researchers from Martin Lysak's research group, in collaboration with scientists from the Institute of Vegetables and Flowers in Beijing, China, sequenced and assembled the ~330-Mb genome of the ephmeral Heliophila variabilis. The work was now published in Plant Journal. Two pairs of differently fractionated subgenomes suggest an allo-octoploid origin of Heliophila genomes – the condition in which eight complete sets of chromosomes occur in a single cell. The ancestral octoploid genome (2n = 8x = ~60) likely arose by hybridization between two allotetraploids (2n = 4x = ~30) at least 12 million years ago. Rediploidization of the ancestral genome was marked by extensive reorganization of the parental subgenomes, resulting in a reduction in chromosome number to only 11 pairs in H. variabilis. The long-lasting diploidization process camouflaged the octoploid nature of Heliophila genomes, which has now been discovered by whole-genome sequencing. The sequenced H. variabilis represents the first chromosome-scale genome assembly of a meso-octoploid representative of the mustard family.
Analysis of the H. variabilis genome has shown that post-polyploid diploidization plays an important role in ecomorphological divergence and adaptation. Evidence has been found for loss-of-function changes in genes associated with leaf development and early flowering, as well as for over-retention and sub/neofunctionalization of genes involved in pathogen response and chemical defense. The genomic resources of H. variabilis will help elucidate the role of polyploidization and genome diploidization in plant adaptation to hot arid environments and origin of the Cape flora.
Although the South African Cape flora is one of the most remarkable biodiversity hotspots, its high diversity has not been associated with whole-genome duplications. The sequenced genome of H. variabilis challenges the traditional picture of the limited contribution of polyploidy to the diversity of the South African flora and suggests that polyploidization–diploidization cycles may have been more widespread in the flora than previously thought.
PhD project in centromere genomics
Topic title: The “black holes” of plant chromosomes: centromere structure and evolution
Supervisor: prof. Martin Lysak (martin.lysak@ceitec.muni.cz)
Annotation:
Centromeres are crucial for the division of eukaryotic cells, because they are required for attachment to spindle microtubules during the segregation of monocentric chromosomes. Though the role of centromeres is conserved across eukaryotes, they exhibit surprising diversity in sequence, structure, and size within and between species. Until recently, megabase-long arrays of repetitive sequences frequently precluded assembly and detailed comparative analysis of centromeres. Now, improvements in long read sequencing approaches and advances in read alignment and genome assembly enabled telomere-to-telomere (T2T) genome assemblies, and thus, detailed characterization of centromere structure. This PhD project will focus on analysis of centromere structure, interphase organization and evolution in crucifer species (Brassicaceae, the mustard family) on sequence and epigenetic level. Comparisons will include all available T2T assemblies with the aim to identify shared vs specific features of crucifer (peri)centromeres and elucidate drivers and trends of their evolution.
Recommended literature:
Naish M et al. (2022) The genetic and epigenetic landscape of the Arabidopsis centromeres. Science 374: eabi7489. doi: 10.1126/science.abi7489.
Wlodzimierz P et al. (2023) Cycles of satellite and transposon evolution in Arabidopsis centromeres. Nature 618: 557-565. doi: 10.1038/s41586-023-06062-z.
Requirements on candidates:
The ideal candidate will have experience analyzing and interpreting whole-genome sequence data. S/he should have good analytical and programming skills.
Keywords:
comparative plant genomics, plant paleogenomics, epigenetics, bioinformatics, centromere, DNA repeats, chromosome and karyotype evolution, genome assembly, whole-genome duplications
Nature paper: Evolutionary cycles in Arabidopsis centromeres

Centromeres, the DNA sections often found in the middle of chromosomes, display enormous diversity within and between species, despite having the same vital role during cell division across almost the entire tree of life. An international team of researchers has discovered that the variation in centromere DNA regions can be strikingly large even within a single species. The findings, now published in the journal Nature, shed light on the molecular mechanisms of rapid centromere evolution. We contributed to this important study of centromere diversity by cytogenetic cross-validation of sequence data. Congratulations to all co-authors!
The missing maternal genome of camelina was identified
A tetraploid C. neglecta-like genome (2n = 4x = 26) was hypothesized by us to be the likely maternal ancestor of the hexaploid Camelina sativa (Mandáková et al., 2019, Plant Cell). In the Plant J paper we report the chromosome-level structure of the predicted tetraploid parental genome previously classified as C. microcarpa and referred to as C. intermedia. The identification of the missing tetraploid donor genome allowed us to paint a more complete picture of the reticulate evolutionary history of the genus Camelina.
Chloroplast phylogenomics in camelina
The 84 assembled chloroplast genomes were used to infer phylogenetic phylogenetic relationship among Camelina species. The research was published in Horticultural Research.
The fascinating evolutionary story of the Biscutelleae published in MBE!
We show that genome evolution in the crucifer tribe Biscutelleae (c. 60 species, 5 genera) was dominated by pervasive hybridizations and subsequent genome duplications. By using complementary cytogenomics and phylogenomics approaches, we demonstrate that the origin of a monophyletic plant clade can be more complex than a parsimonious assumption of a single whole-genome duplication (WGD) spurring post-polyploid cladogenesis. Recurrent hybridization among the same and/or closely related parental genomes may phylogenetically interlink diploid and polyploid genomes despite the incidence of multiple independent WGDs. Our results elucidate challenges in resolving the contentious relationships within and between land plant lineages with pervasive hybridization and WGDs. The fascinating evolutionary jigsaw puzzle was published by Molecular Biology and Evolution.
Martin Lysak was awarded MUNI Scientist Award 2020

Selected scientists from Masaryk University were awarded for their outstanding research results and significant achievements in the field of research from 2019 to 2020. More information can be found here.
Vaccant positions
Teri received the GACR President's Award!
The prize is awarded for excellent results in basic research projects that had been supported by the Czech Science Foundation (GACR). Teri was awarded for her research on Camelina (false flax) genomes published in Plant Cell. Congratulations!
Plant Cell paper: Collinear Chromosomes and Shifting Centromeres in the Arabideae
Our paper on genome evolution and frequent centromere repositioning in one of the Brassicaceae clades, Arabideae, was published in Plant Cell! Our study documented frequent centromere shifts in the largest species set within a single monophyletic plant clade. The paper was highlighted by an In Brief article by Jen Mach.
Genome history of camelina is emerging from the shadows. Plant Cell paper.

Sequencing of the camelina (Camelina sativa) genome revealed that the genome consists of three parental subgenomes. However, how the hexaploid camelina genome originated and how the three subgenomes are related to other genomes in the genus Camelina remained unknown. We tackled these questions by using comparative chromosome painting, genomic in situ hybridization, and multi-gene phylogenetic analyses. We aimed to identify the most probable parental genomes of the hexaploid camelina and moreover to understand how its genome is related to genomes of other Camelina species.
Genomes of diploid camelinas (C. hispida, n = 7 chromosomes; C. laxa, n = 6; and C. neglecta, n = 6) originated from an ancestral n = 7 genome. The allotetraploid C. rumelica genome (n = 13, N6H genome) arose from hybridization between diploids C. neglecta (n = 6, N6) and C. hispida (n = 7, H), and the N subgenome has been substantially reshuffled by chromosomal rearrangements. The allohexaploid genomes of C. sativa (n = 20, N6N7H) originated through hybridization between an auto-allotetraploid C. neglecta-like genome (n = 13, N6N7) and C. hispida (n = 7, H), and the three subgenomes remained overall stable since the genome merger. Remarkably, the ancestral and diploid Camelina genomes were shaped by complex chromosome shattering, resembling similar events associated with human disorders.
The Camelina story was published in Plant Cell!
The paper was highlighted by a Plant Cell editorial by Jen Mach: Camelina: A History of Polyploidy, Chromosome Shattering, and Recovery.
"Our promiscuous bittercresses" highlighted in Annals of Botany
Mike Chester (Royal Botanic Gardens, Kew) posted a comment on our paper published in Annals of Botany earlier this year (Mandáková T et al., The story of promiscuous crucifers: origin and genome evolution of an invasive species, <i>Cardamine occulta</i> (Brassicaceae), and its relatives.)
Special Issue "Chromosomal Evolution in Plants"
Martin Lysak and Hanna Schneeweiss (University of Vienna) are guest editors of a research topic in Frontiers in Plant Science entitled Chromosomal Evolution in Plants.
About this Research Topic
Plant karyotypes display a great diversity in chromosome number and morphology, as well as DNA amount and composition. Chromosomal evolution, one of the driving forces underlaying diversification and speciation in plants, is usually complex and employs various mechanisms. Although chromosome number and karyotype structure are still the most frequently recorded cytological characters, last decades have brought a range of new tools that allow for better insights into chromosomal and genome evolution in plants. The massively parallel sequencing technology (next-generation sequencing, NGS) has in last few years revolutionized nearly all biological disciplines. Genome skimming using NGS and whole-genome sequences opened up new horizons for modern evolutionary cytogenetics, or more accurately, cytogenomics. It therefore became obvious that plant evolutionary cytogenomics can benefit from whole-genome sequences, and comparative plant genomics can be associated to physical localization of DNA sequences on chromosomes, improving whole-genome and chromosome-level assemblies. Thus, we are now able to take full advantage of the modern approaches to finally tackle the long-standing questions in chromosome biology and genome evolution in any plant species group.
This Research Topic aims to bring together a collection of articles addressing important questions related to genome and chromosomal evolution across the green plant phylogeny including flowering plants, gymnosperms, ferns, bryophytes and algae. Studies presenting novel and exciting data on genome and chromosomal evolution, as well as new methodological tools, are welcome. The goal is to advance our understanding of plant karyotype evolution and its contribution to diversification and speciation, and allow for exchange of data, ideas, and hypotheses obtained making full advantage of new methodological approaches.
Specifically, we encourage submission of Original Research, Reviews, Mini Reviews, Methods, Perspectives, and Opinions covering the following topics:
- Reconstructing pathways of karyotype and genome evolution
- Paleogenomics and paleogenomes
- Polyploidy and post-polyploid genome diploidization
- Dysploid chromosomal changes
- Structure of plant chromosomes
- Impact of interphase chromosomes organization on genome evolution
- Repeatome and genome size evolution
- The role of chromosomal rearrangements in plant speciation
Keywords: Plant Karyotype, Plant Genome Evolution, Cytogenetics, Cytogenomics, Chromosomal Evolution

The structure of horseradish and watercress genomes uncovered!

In a study published in Plant Physiology, we showed that the tetraploid horseradish and watercress genomes are almost identical structurally (differentiated only by a 2.4-Mb chromosome translocation), and that presumably both originated by autopolyploidization. Both genomes closely resemble bittercress (Cardamine) genomes from the same tribe. This analysis represents a first stepping stone for the future whole-genome sequencing efforts and genetic improvement of both crop species. The paper was highlighted on CEITEC's home page: https://www.ceitec.eu/scientists-have-mapped-the-horseradish-and-watercress-genome/t10039.
The Aquilegia paper higlighted at CEITEC's website

Our joint paper about the eleven sequenced columbine genomes and one odd chromosome published in eLife recently was highlighted in the form of a press release at CEITEC's website.
The Ricotia story published in Plant Journal - recurrent origin of a fusion chromosome!
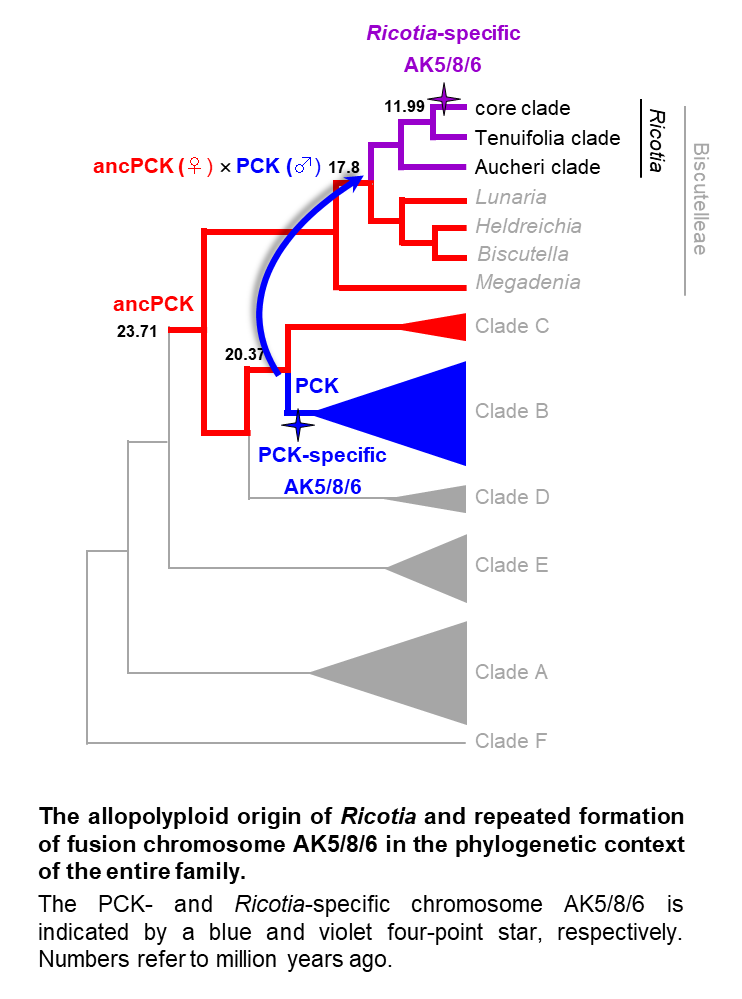
The ancestral Ricotia genome was formed by a rare inter-clade hybridization event, followed by cladogenesis accompanied by three decreases in chromosome number (descending dysploidies). The last dysploidy was mediated by a unique chromosomal rearrangement, proving that structurally identical or similar fusion chromosomes may be formed in land plants from the same precursor chromosomes by different translocation mechanisms repeatedly after several million years.
Teri Mandáková awarded for outstanding scientific results

Teri was award for extraordinary research results achieved by scientists in the age group under 35 from the Rector of Masaryk University. Congratulations!
Post-polyploid diploidization and diversification through dysploid changes
Our review on the role of chromosomal rearrangements and descending dysploidies in post-polyploid diploidization in plants was published as a part of the special issue Genome studies and molecular genetics 2018 of Current Opin Plant Biol (https://www.sciencedirect.com/journal/current-opinion-in-plant-biology/v...)!
The exciting story of an endemic Australian crucifer clade was published in Molecular Ecology

These results are among the first to demonstrate multispeed genome evolution in taxa descending from a common allopolyploid ancestor. Clade-specific post-polyploid diploidization can operate at different rates and efficacies and can be tentatively linked to life histories and the extent of taxonomic diversity.
Our paper on mesopolyploid whole-genome duplications selected as a Featured Article in Plant Journal

Our paper reporting on concealed whole-genome duplications in several Brassicaceae tribes and genera was selected as a Featured Article by the editors of Plant Journal and advertised by the cover illustrating the mesohexaploid origin of the South African tribe Heliophileae (c. 100 species).
We were awarded an Inter-Excellence grant to work on tribe Boechereae
We were successful with our application for a three-year Inter-Excellence research grant to work on genome evolution of tribe Boechereae, endemic to North America. This is a joint project with the laboratory of prof. Tom Mitchell-Olds at the Duke University.
A chromosome inversion affects multiple ecologically important traits in Boechera

What is the role of inversions in adaptation and speciation? New data on the adaptive role of a young paracentric inversion were gained in the study led by Tom Mitchell-Olds (Duke University) and published in the April issue of Nature Ecology and Evolution. This paper is a follow-up of our Boechera study published in Plant Journal 82, 785-793 (2015).
ICPHB2016, 11-14 May 2016, Rovinj, Croatia
Teri gave an invited lecture on "Multiple patterns of genome evolution in the Brassicaceae: a lesson from the polyploid-rich genus Cardamine" at the International Conference on Polyploidy, Hybridization and Biodiversity in Rovinj, Croatia.
Ancestral building blocks of crucifer genomes revisited
We revisited the concept of the Ancestral Crucifer Karyotype (ACK) and the definition of a revised set of 22 conserved genomic blocks across the Brassicaceae family including Arabidopsis and crop Brassicas. We reviewed how the ACK has been utilized for the analysis of a remarkable thirty-five crucifer genomes during the last 10 years. This work was published in Current Opinion in Plant Biology.
chromDraw graphic tool for karyotype visualization
Basic features of the chromDraw tool are described in our recent paper published in Chromosome Research.
TheBoechera paper and Lysak lab highlighted in Lab Times's journal club
Plant Cytogenomics in Brno, Czech Republic: Treading in Mendel’s Genomic Footsteps
Insight into evolution of the Noccaea /Thlaspi caerulescens genome

In the September issue of Plant Physiology, we describe the comparative genome structure of the Alpine Pennycress (Noccaea or Thlaspi caerulescens) and its evolution from an ancestral genome. We show that genome evolution in this important model species involved an unusually high number of pericentric inversions, which could have facilitated the evolution of enhanced metal homeostasis gene expression, a known hallmark of metal hyperaccumulation.
Our paper on chromosome evolution in Boechera highlighted in Trends in Plant Science

A Spotlight article written by Marcus Koch (University of Heidelberg) can be downloaded from the link below.
Botany 2015 - colloquium Recent advances in phylogeny and systematics of Brassicaceae !

BRASSICACEAE @ Botany 2015 - Edmonton, Alberta - July 25 - 29, 2015
A colloquium on recent advances in phylogenetics, systematics and genome evolution of Brassicaceae will bring together leading experts in the field. Among our speakers are Ihsan Al-Shehbaz, Ivalu Cacho, Hong Ma, Karol Marhold, Marcus Koch, and Eric Schranz.
Organizers: K. Marhold and M. A. Lysak
Structure and origin of aberrant chromosomes in apomictic Boechera species revealed

Mandáková et al. (2015) Karyotype evolution in apomictic Boechera and the origin of the aberrant chromosomes. Plant Journal 82: 785–793.
The genome of Alpine Rockcress

Willing et al. Genome expansion of Arabis alpina linked with retrotransposition and reduced symmetric DNA methylation. Nature Plants (doi:10.1038/nplants.2014.23)
An international team led by Georg Coupland and Korbinian Schneeberger (MPI for Plant Breeding Research, Cologne) published a de novo assembly for the 375 Mb genome of A. alpina. This paper also reports on the structure and evolution of the eight chromosomes of this important perennial model plant. The A. alpina genome is the first sequenced genome from the largest tribe of Brassicaceae (the Arabideae contain c. 487 species in 17 genera).
Genome evolution and spatiotemporal diversity in the tribe Arabideae

We were recently awarded a three-year project from the Czech Science Foundation to investigate karyotype and genome evolution in the Arabideae.
The tribe Arabideae is the largest Brassicaceae tribe with >487 species; many Arabis and Draba species being wide-spread and some cultivated as ornamental plants. Arabideae diversified into several clades with contrasting phylogeographic history represents a superb system to analyze plant genome evolution. We aim to describe and reconstruct patterns of genome reorganization and repeat evolution by comparative cytogenomic and epigenetic methods, and by next-generation sequencing. 01/2015 - 12/2017.
Investigating genomes of Icelandic crucifer species

The EEA project aims to elucidate genome structure and evolution in selected Icelandic crucifer genera (e.g., Cardamine, Draba and Subularia).
A joint project between the Lysak lab and the group of prof. Kesara Anamthawat-Jónsson (Unversity of Iceland). 04/2015 - 05/2016.
More information about the project can be found here.
Terminal and interstitial telomeric repeats

Majerova E., Mandáková T., Vu G.T.H., Fajkus J., Lysak M.A., Fojtova M. 2014. Epigenetic character of telomeric chromatin in model plants with distinct localization of telomeric repeats. Frontiers in Plant Science 5:593. [PDF]